In Vitro Dissolution Rate Testing
Dissolution testing was first introduced in the early 1960s as the problems associated with the biological availability of drugs became more apparent. Bioavailability rate testing in industry became increasingly widespread as a means of distinguishing between two competitive drugs that were otherwise pharmaceutically identical in their physical properties, according to all other tests at that time.
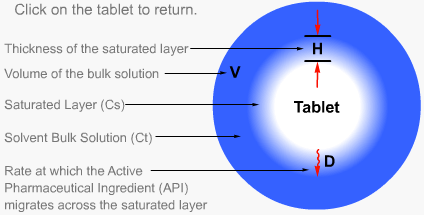
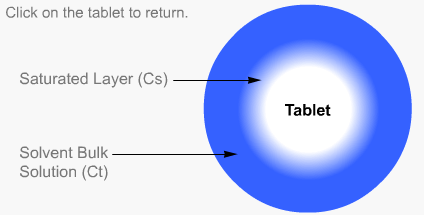
Intrinsic dissolution rate is described as the time taken to dissolve a specified mass of Active Pharmaceutical Ingredient (API) with a constant surface area in a solvent. It is characteristic of each solid compound in a given solvent under fixed experimental conditions and is useful in predicting probable bioavailability problems due to dissolution rate.
The value is generally expressed as mg/min/cm². Intrinsic rates of 1.0 mg/min/cm² have negligible problems with dissolution rate limitations.
However, rates of less than 0.1mg/min/cm² suggest that the drug is dissolution rate limited.
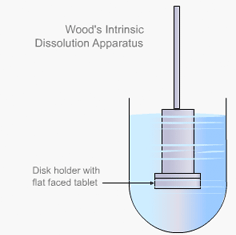
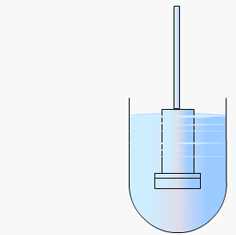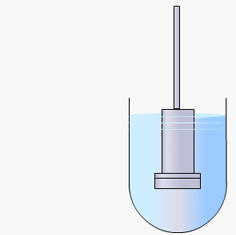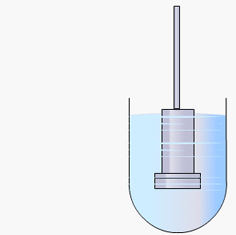
Particulate dissolution rate is measured by observing the time taken for a suspension of the API to dissolve in a fixed amount of solvent without fixed control of the dose surface area. It is used to study the influence of particle size, surface area and excipients upon the active agent.
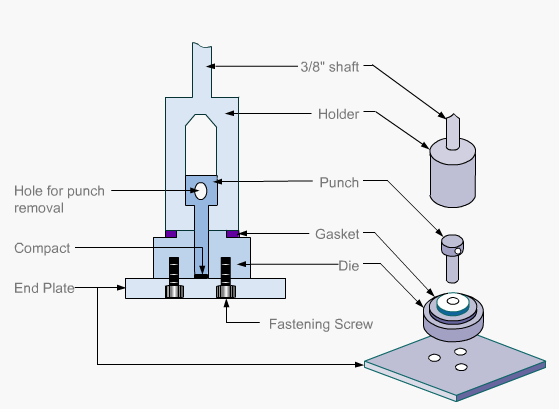
Particulate dissolution rate is used, along with intrinsic dissolution, to measure the dissolution rates of chemical compounds. Typically, a weighed amount of powdered sample is added to the dissolution medium in a constant agitation system.

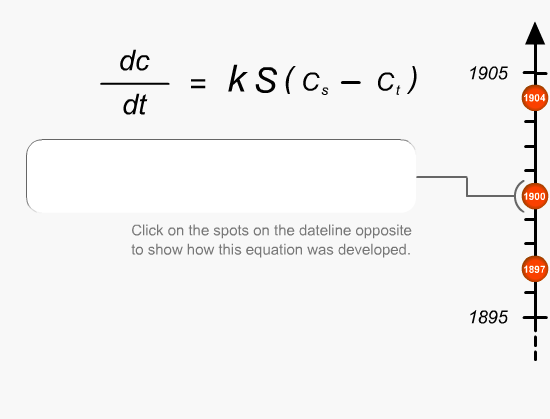



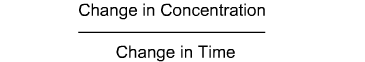
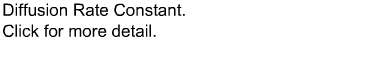
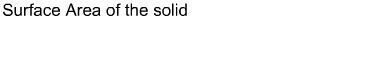





Equations
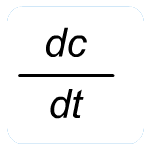
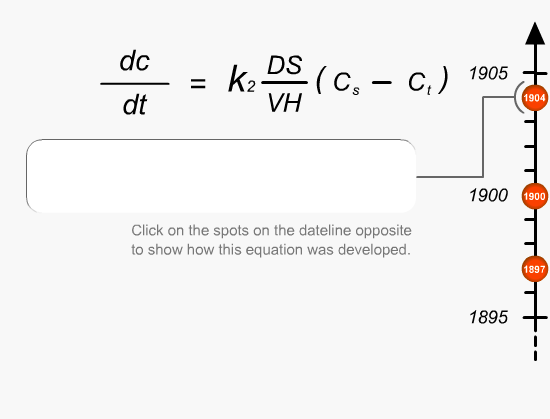
This is the first fundamental equation established by Noyes and Whitney to determine the rate of dissolution. The dissolution constant is unique for each material under test and can be determined by measuring the dissolution rate of the material in a defined solvent while keeping the surface area of the material constant at a fixed temperature.Equations
Brunner and Tollockzo modified the Noyes and Whitney equation to predict the dissolution rate constant under sink conditions when the volume and surface area is kept constant. Sink conditions can be approximated by keeping the volume of the solvent five to ten times larger than the saturation volume of the medium. They are one of the main experimental parameters that need to be controlled during dissolution testing.Equations
Nernst and Brunner slightly modified the Brunner and Tollockzo Equation to produce a more comprehensive description of the relationship. The elements that comprise the equation can be varied by a number of different external factors (eg. paddle speed, temperature and vibration). Slight changes in any of these factors can actually change the rate at which the product passes into solution.