

Acceptance Criteria for Apparatus 1 and 2
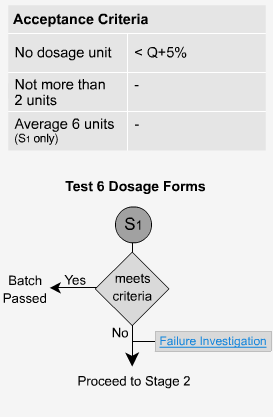
- What is Q?
Immediate
ReleaseDelayed
ReleaseExtended
ReleaseTransdermal
Dosage Forms
Most immediate release products have a particular specification - a certain quantity of material should be released in a certain period of time. This quantity is expressed as a percentage of label claim, that amount given on a dosage form which specifies how much drug is in each tablet or capsule.
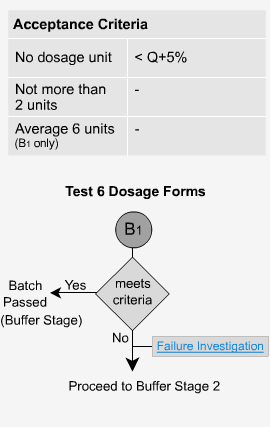
The quantity of Q is the amount of dissolved active ingredient specified in the individual monograph expressed as a percentage of the label claim.
The typical Q specifications for the amount of active ingredient dissolved are in the range of 70% to 80%. A Q value in excess of 80% is not generally used.
The development of the test times and specifications are usually established on the basis of an evaluation of dissolution profile data.
- Overview
- Stage 1
- Stage 2
- Stage 3
Immediate Release Overview
Immediate Release formulations are the most common type of dosage form and are designed to dissolve in the acidic conditions of the stomach. To simulate the gastric environment, 0.1M hydrochloric acid is commonly used as the medium and the dissolution test usually takes 30 - 60 minutes to perform. Immediate release tests 45 minutes or longer generally require two sampling time points
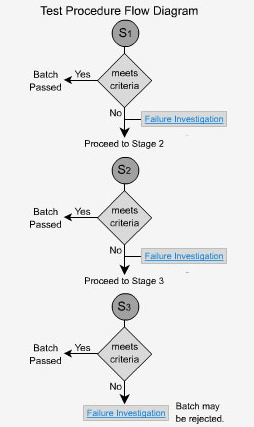
Immediate Release - S1
Per USP: Each unit tested must be not less than Q+5% to pass (eg. if a monograph or method states Q = 80%, each tablet must not be less than 85% to pass).
Immediate Release - S2
If the criteria for stage 1 are not met then 6 additional dosage forms are tested. The average of the results from both stage 1 and stage 2, a total of 12 dosage forms in all, is calculated and the value must be greater than or equal to Q. No individual unit should have a value less than Q-15%. In the previous example where Q = 80%, no unit should be less than 65% to pass stage 2.

Immediate Release - S3
An additional 12 units are tested here and the average of all 24 tablets from all three stages is taken. This value must not be less than the initial value of Q. In addition, no more than two units can have values less than Q-15% and no tablet can have a value less than Q-25%. If the samples fail at this stage, the whole batch is investigated and possibly rejected. In the previous example where Q = 80%, no two units would be less than 65% and no unit would be less than 55% to pass stage 3.

- Overview
- Acid Stage 1
- Acid Stage 2
- Acid Stage 3
Delayed Release Acid Stage Overview
Delayed Release (enteric-coated) products fall under the USP General Chapter 711, Dissolution. Delayed release forms are designed to pass through the gastric region and not dissolve in the stomach by virtue of a protective coating. The dissolution test is divided into stages to simulate these conditions - an initial acid stage and an alkaline buffer stage.
In the acid stage, 0.1 N hydrochloric acid or simulated gastric fluid is used to mimic the conditions of the stomach. The dosage forms are left for two hours ± 2%. After samples are removed from the end of the acid stage the media is changed or adjusted to buffered conditions and the test continues.
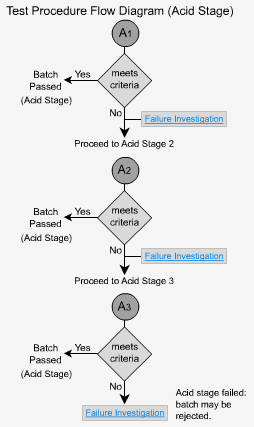
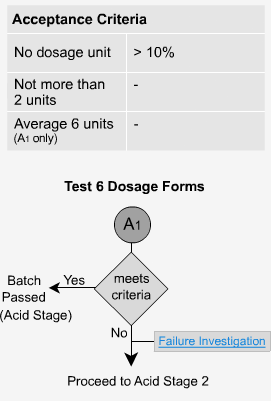
Delayed Release - A1
Six dosage forms are tested and no individual unit should exceed 10% dissolved. This is because the form is enteric coated and should not dissolve.
Delayed Release - A2
If the criteria for stage 1 is not met, then six more dosage forms are tested.
The average of all 12 (A1 + A2) is not more than 10% dissolved and no unit is greater than 25% dissolved. Once again, the low value should indicate that the samples have only dissolved minimally, or not at all.

Delayed Release - A3
If the dosage forms do not pass the second stage, an additional 12 units must be tested.
The average of all 24 units (A1 + A2 + A3) is not more than 10% dissolved and no unit is greater than 25% dissolved. Once again, low values are to be expected.

- Overview
- Buffer Stage 1
- Buffer Stage 2
- Buffer Stage 3
Delayed Release Buffer Stage Overview
At the end of the acid stage, the sample must be withdrawn and media added/ adjusted within FIVE minutes. Typically, add 250 mL of 0.20-M tribasic sodium phosphate buffer at 37°C, adjust the pH to 6.8 ± 0.05 while stirring at the specified rate. The dissolution test continues for 45 minutes (or per monograph specifications).
Temperature conditions at 37 ± 0.5°C must be maintained throughout the test..
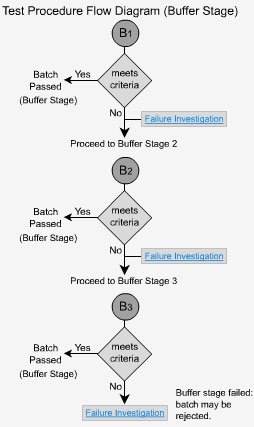
Delayed Release - B1
Samples are taken at the end of the buffer stage, at which point the dosage form should have dissolved. The initial value of Q is calculated as being the total of both the acid and buffer stages. The value of Q is 75% dissolved unless otherwise specified in the individual monograph. The extended release forms should now behave as immediate release forms and so the evaluation of Q is calculated accordingly. Each unit tested must be not less than Q+5% to pass (eg. if Q = 80%, each tablet must not be less than 85% to pass).
Delayed Release - B2
If the criteria for buffer stage 1 is not met, then six additional dosage forms are tested.
The average of the results from both stages (B1 + B2), a total of 12 dosage forms in all, is calculated and the value must be greater than or equal to Q. No individual unit should have a value less than Q-15%. In our previous example, when Q = 80%, no unit should be less than 65% to pass stage 2.

Delayed Release - B3
If the dosage forms do not pass the second stage, then 12 additional units are tested.
The average of all 24 units (B1 + B2 + B3) is calculated. The value must not be less than the initial value of Q. In addition, no more than two units can have values of < Q-15% and no tablet can have a value of < Q-25%.
If the samples fail at this stage, the entire batch is investigated and possibly rejected. In our previous example where Q = 80%, no two units would be less than 65% and no unit would be less than 55% to pass stage 3.

- Overview
- Level 1
- Level 2
- Level 3
Extended Release Overview
Extended Release products fall under the USP General Chapter 711, Dissolution. Extended release dosage forms are designed to release the active ingredient over a longer period of time than other dosage forms. During testing, sample aliquots are taken at monograph specified time intervals ±2%. Generally, three sample time periods may be specified with the monograph providing a range of dissolved ingredients for each.
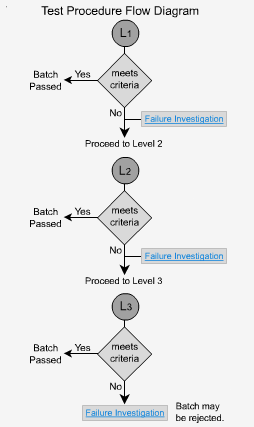
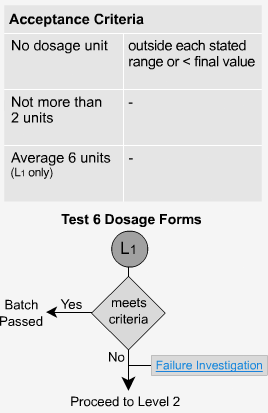
Extended Release - L1
In order to pass the initial level, sample test results of six dosage forms must fall between the upper and lower limits of the specified range for each time period. No unit should be less than the stated amount at the final test time.
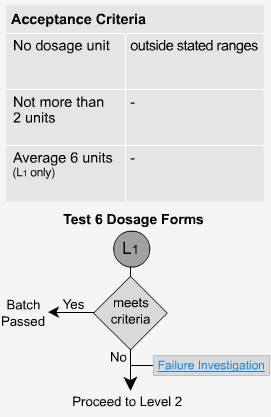
Extended Release - L2
If the dosage forms fail level 1, then 6 additonal samples are analyzed. The average of all 12 units (L1 + L2) must fall within the stated range for each time period and be not less than the stated amount at the final test time. No single unit should be more than 10% of the labeled content outside each of the stated ranges and no single unit should be more than 10% of the labeled content below the stated amount at the final test time.
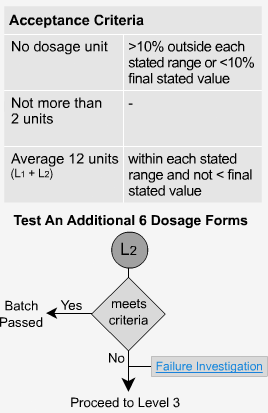
Extended Release - L3
If the samples fail level 2, then 12 more samples must be analyzed. The average of all 24 units (L1 + L2 + L3) should fall within each time period's specified range and be not < the stated amount at the final test time. Not more than 2 units should be >10% outside each of the stated ranges or 10% of the labelled content below the stated amount at the final test time. No unit should be greater than 20% of labeled content outside each of the stated ranges or greater than 20% of the labeled content below the stated amount at the final test time, or the whole batch may have to be rejected.

- Overview
- Level 1
- Level 2
- Level 3
Transdermal Dosage Forms Overview
Transdermal dosage forms were developed in the late 70s and early 80s during which time they became extremely popular. They are not administered orally but as a patch on the skin and are a good way of providing a continuous amount of active drug substance in a linear release profile as opposed to the familiar release curve typical of oral dosage forms.
Three sample time periods are typically specified with the monograph indicating a range of dissolved ingredients for each. These forms are typically tested using modifications to the classic apparatus 1 and 2 setups.
Transdermal Dosage Forms - L1
Of the six dosage forms tested, all units should be within the specified range of released material for each time period.
Transdermal Dosage Forms - L2
If the dosage forms do not meet level 1 criteria, then 6 additional samples are analyzed. The average of all 12 units (L1 + L2) must fall within the stated ranges for each sample time period and no single unit should be outside the stated range by greater than 10% of the average of the stated range.

Transdermal Dosage Forms - L3
If the samples do not meet level 2 criteria, then an additional 12 samples must be analyzed. The average of all 24 units (L1 + L2 + L3) should fall within the specified ranges for each sample time period. No greater than 2 units should be outside the stated ranges by greater than 10% of the average of each range and no unit is outside the stated ranges by greater than 20% of the average of each range.
